Opis:
Hipercholesterolemia i dyslipidemia mieszana. Produkt leczniczy jest wskazany do stosowania u dorosłych z pierwotną hipercholesterolemią (rodzinną heterozygotyczną i nierodzinną) lub mieszaną dyslipidemią jako uzupełnienie diety: w skojarzeniu ze statyną lub statyną i innymi lekami hipolipemizującymi u pacjentów, u których nie jest możliwe osiągnięcie docelowego stężenia cholesterolu LDL przy zastosowaniu statyny w najwyższej tolerowanej dawce, albo; w monoterapii lub w skojarzeniu z innymi lekami hipolipemizującymi u pacjentów nietolerujących statyn, albo u których stosowanie statyn jest przeciwwskazane. Postać homozygotyczna rodzinnej hipercholesterolemii. Produkt leczniczy jest wskazany do stosowania w skojarzeniu z innymi lekami hipolipemizującymi u dorosłych i młodzieży w wieku co najmniej 12 lat z homozygotyczną postaciąhipercholesterolemii rodzinnej. Rozpoznana miażdżycowa choroba układu sercowo-naczyniowego. Produkt leczniczy jest wskazany do stosowania u osób dorosłych z rozpoznaną miażdżycowąchorobą układu sercowo-naczyniowego (zawał mięśnia sercowego, udar mózgu lub choroba tętnic obwodowych) w celu zmniejszenia ryzyka chorób układu sercowo-naczyniowego poprzez zmniejszenie stężenia cholesterolu LDL, jako uzupełnienie skorygowania innych czynników ryzyka: w skojarzeniu z maks. tolerowaną dawką statyny lub bez innych terapii zmniejszających stężenie lipidów lub, samodzielnie lub w połączeniu z innymi terapiami zmniejszającymi stężenie lipidów u pacjentów z nietolerancją statyn, lub u których stosowanie statyn jest przeciwwskazane. Wyniki badań dotyczących wpływu produktu na stężenie LDL-C, incydenty sercowo-naczyniowe i badane populacje.
Przed rozpoczęciem stosowania produktu leczniczego należy wykluczyć wtórne przyczyny hiperlipidemii lub mieszanej dyslipidemii (np. zespół nerczycowy, niedoczynność tarczycy). Hipercholesterolemia pierwotna i dyslipidemia mieszana u dorosłych: zalecana dawka produktu leczniczego to 140 mg raz/2 tyg. lub 420 mg raz w m-cu; obydwie dawki są klinicznie równoważne. Homozygotyczna postać hipercholesterolemii rodzinnej u dorosłych i młodzieży w wieku co najmniej 12 lat: zalecana dawka początkowa produktu leczniczego to 420 mg raz w m-cu. Po 12 m-cach leczenia, częstość dawkowania może być stopniowo zwiększana do 420 mg raz na 2 tyg., jeśli nie została uzyskana odpowiedź istotna klinicznie. U pacjentów, u których wykonywana jest afereza, można rozpocząć leczenie dawką 420 mg podawaną raz na 2 tyg. w taki sposób, aby schemat dawkowania dopasować do harmonogramu zabiegów aferezy. Pacjenci z zaburzeniami czynności nerek. U pacjentów z łagodnymi lub umiarkowanymi zaburzeniami czynności nerek nie jest konieczna modyfikacja dawkowania, patrz sekcja Ostrzeżenia specjalne/Środki ostrożności - pacjenci z ciężkimi zaburzeniami czynności nerek (eGFR <30 ml/min/1,73 m2). U pacjentów z łagodnymi zaburzeniami czynności wątroby nie jest konieczna modyfikacja dawkowania, patrz sekcja Ostrzeżenia specjalne/Środki ostrożności - pacjenci z umiarkowanymi i ciężkimi zaburzeniami czynności wątroby. Pacjenci w podeszłym wieku (≥65 lat): u pacjentów w podeszłym wieku nie jest konieczna modyfikacja dawkowania. Dzieci i młodzież. Nie określono bezpieczeństwa stosowania ani skuteczności produktu leczniczego u dzieci <18 lat w leczeniu pierwotnej hipercholesterolemii i mieszanej dyslipidemii. Nie ma dostępnych danych. Nie określono bezpieczeństwa stosowania ani skuteczności produktu leczniczego u dzieci w wieku poniżej 12 lat w leczeniu homozygotycznej postaci rodzinnej hipercholesterolemii. Nie ma dostępnych danych.
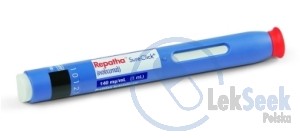
Podanie podskórne. Produkt leczniczy przeznaczony jest do podawania we wstrzyknięciach podskórnych w powłoki jamy brzusznej, w udo lub górną część ramienia. Miejsce wstrzyknięcia należy zmieniać sekwencyjnie. Nie należy wykonywać wstrzyknięć w miejscach, w których skóra jest tkliwa, zasiniona, zaczerwieniona lub stwardniała. Produktu leczniczego nie wolno podawać dożylnie ani domięśniowo. Dawkę 420 mg należy podawać raz w m-cu lub raz na 2 tyg., wstrzykując lek kolejno z 3 wstrzykiwaczy półautomatycznych napełnionych w ciągu 30 minut. Produkt leczniczy jest przeznaczony do samodzielnego wstrzykiwania przez pacjenta po odpowiednim przeszkoleniu. Produkt leczniczy może być również podawany przez osobę odpowiednio przeszkoloną w zakresie techniki wstrzykiwania produktu. Każdy wstrzykiwacz półautomatyczny napełniony przeznaczony jest wyłącznie do jednorazowego wykorzystania.
Nadwrażliwość na substancję czynną lub na którąkolwiek substancję pomocniczą.
Pacjenci z ciężkimi zaburzeniami czynności nerek (określonymi jako eGFR < 30 ml/min/1,73 m2) nie byli badani. Produkt leczniczy powinien być stosowany ostrożnie u pacjentów z ciężkimi zaburzeniami czynności nerek. U pacjentów z umiarkowanymi zaburzeniami czynności wątroby obserwowano, że zmniejszenie całkowitej ekspozycji na ewolokumab może prowadzić do zmniejszenia efektu redukcji stężenia cholesterolu LDL. W związku z czym, może być uzasadnione ścisłe monitorowanie tych pacjentów. Pacjenci z ciężkimi zaburzeniami czynności wątroby (C w skali Childa-Pugha) nie byli badani. Produkt leczniczy powinien być stosowany ostrożnie u pacjentów z ciężkimi zaburzeniami czynności wątroby. Osłonka na igłę we wstrzykiwaczu półautomatycznym napełnionym wykonana jest z suchej naturalnej gumy (pochodnej lateksu) i może powodować wystąpienie reakcji alergicznych. Ten produkt leczniczy zawiera mniej niż 1 mmol (23 mg) sodu w jednej dawce, co oznacza, że zasadniczo jest „wolny od sodu”. Preparat nie ma wpływu na zdolność prowadzenia pojazdów i obsługiwania maszyn.
Nie przeprowadzono formalnych badań dotyczących interakcji produktu leczniczego z innymi lekami. Interakcje farmakokinetyczne pomiędzy statynami a ewolokumabem oceniano w badaniach klinicznych prowadzonych z zastosowaniem produktu leczniczego. U pacjentów, którym podawano jednocześnie statyny, obserwowano zwiększenie wartości klirensu ewolokumabu o około 20%. Do zwiększenia wartości klirensu częściowo przyczyniają się statyny powodujące zwiększenie stężenia konwertazy białkowej subtylizyny/keksyny typu 9 (ang. Proprotein Convertase Subtilisin/Kexin Type 9, PCSK9), co jednak nie wpływa w sposób niekorzystny na farmakodynamiczne działanie ewolokumabu na lipidy. Nie jest konieczna modyfikacja dawki statyny podczas stosowania w skojarzeniu z produktem leczniczym. Nie badano interakcji farmakokinetycznych i farmakodynamicznych pomiędzy produktem leczniczym i lekami hipolipemizującymi innymi niż statyny i ezetymib.
Nie ma danych dotyczących stosowania produktu leczniczego u kobiet w ciąży lub ilość tych danych jest ograniczona. Wyniki badań na zwierzętach nie wskazują na bezpośredni ani pośredni toksyczny wpływ na rozród. Produktu leczniczego nie należy stosować w czasie ciąży, chyba że kobieta wymaga leczenia ewolokumabem ze względu na stan kliniczny. Nie wiadomo, czy ewolokumab przenika do mleka kobiecego. Nie można wykluczyć zagrożenia dla noworodków lub niemowląt karmionych piersią. Należy podjąć decyzję, czy przerwać karmienie piersią, czy zrezygnować ze stosowania produktu leczniczego lub przerwać terapię tym produktem, po rozważeniu korzyści dla dziecka związanych z karmieniem piersią i korzyści dla matki wynikających z leczenia. Nie ma dostępnych danych dotyczących wpływu ewolokumabu na płodność u ludzi. W badaniach na zwierzętach nie wykazano wpływu na punkty końcowe dotyczące płodności na podstawie pola pod krzywą zależności stężenia od czasu (AUC), gdy ekspozycja (AUC) była dużo większa w porównaniu z ekspozycją pacjentów otrzymujących ewolokumab w dawce 420 mg raz w m-cu.
Najczęściej zgłaszanymi działaniami niepożądanymi w trakcie głównych badań z udziałem pacjentów z pierwotną hipercholesterolemią i mieszaną dyslipidemią podczas stosowania zalecanych dawek były: zapalenie błony śluzowej nosa i gardła (4,8%), infekcje górnych dróg oddechowych (3,2%), ból pleców (3,1%), bóle stawów (2,2%), grypa (2,3%) i nudności (2,1%). Profil bezpieczeństwa w populacji pacjentów z homozygotyczną postacią rodzinnej hipercholesterolemii był spójny z profilem bezpieczeństwa wykazanym w populacji pacjentów z pierwotną hipercholesterolemią i mieszaną dyslipidemią. Działania niepożądane zgłoszone w kluczowych badaniach klinicznych z grupą kontrolną u pacjentów z pierwotną hipercholesterolemią i mieszaną dyslipidemią oraz homozygotyczną postacią rodzinnej hipercholesterolemii. Zakażenia i zarażenia pasożytnicze: (często) zapalenie błony śluzowej nosa i gardła, infekcje górnych dróg oddechowych. Zaburzenia układu immunologicznego: (często) wysypka; (niezbyt często) pokrzywka. Zaburzenia żołądka i jelit: (często) nudności. Zaburzenia mięśniowo-szkieletowe i tkanki łącznej: (często) ból pleców, bóle stawów. Zaburzenia ogólne i stany w miejscu podania: (często) reakcje w miejscu wstrzyknięcia. Do najczęściej występujących działań w miejscu wstrzyknięcia należały: rumień w miejscu wstrzyknięcia, ból w miejscu wstrzyknięcia i zasinienie w miejscu wstrzyknięcia. Doświadczenie związane ze stosowaniem produktu leczniczego u dzieci i młodzieży jest ograniczone. Do badań klinicznych włączono czternastu pacjentów w wieku ≥12 do < 18 lat z homozygotyczną postacią rodzinnej hipercholesterolemii. Nie wykazano różnic w ocenie bezpieczeństwa u młodzieży i dorosłych pacjentów z homozygotyczną postacią rodzinnej hipercholesterolemii. Nie określono bezpieczeństwa stosowania ani skuteczności produktu leczniczego u dzieci i młodzieży z pierwotną hipercholesterolemią i mieszaną dyslipidemią. Chociaż nie obserwowano żadnych problemów dotyczących bezpieczeństwa, dane w podgrupie wiekowej powyżej 75 lat są ograniczone. Spośród wszystkich 6026 pacjentów biorących udział w badaniach klinicznych produktu leczniczego, 1779 (30%) pacjentów było w wieku ≥65 lat, a 223 (4%) w wieku ≥75 lat. Nie obserwowano zasadniczych różnic w zakresie bezpieczeństwa i skuteczności pomiędzy tymi i młodszymi pacjentami. W badaniach klinicznych dodatnie wyniki testów na obecność przeciwciał wiążących uzyskano u 0,1% pacjentów (7 spośród 4846 pacjentów z pierwotną hipercholesterolemią i mieszaną dyslipidemią oraz u żadnego spośród 80 pacjentów z homozygotyczną postacią rodzinnej hipercholesterolemii), którzy otrzymali co najmniej 1 dawkę produktu leczniczego (u 4 spośród tych pacjentów obecność przeciwciał była przemijająca). U pacjentów, u których stwierdzono przeciwciała wiążące w surowicy krwi, wykonano dodatkowo testy na obecność przeciwciał neutralizujących, ale nie wykazano ich u żadnej z tych osób. Obecność przeciwciał wiążących skierowanych przeciwko ewolokumabowi nie miała wpływu na profil farmakokinetyczny, odpowiedź kliniczną ani bezpieczeństwo produktu leczniczego..
W badaniach na zwierzętach nie odnotowano żadnych działań niepożądanych, gdy ekspozycja osiągnęła wartość do 300-krotnie większą niż ekspozycja pacjentów otrzymujących produkt leczniczy w dawce 420 mg raz w m-cu. Nie ma swoistego leczenia po przedawkowaniu produktu leczniczego. W przypadku przedawkowania, należy zastosować leczenie objawowe i, w razie konieczności, wdrożyć postępowanie podtrzymujące.
Ewolokumab wiąże się wybiórczo z PCSK9. W ten sposób uniemożliwia wiązanie krążącej PCSK9 z receptorem dla lipoprotein o niskiej gęstości (ang. low density lipoprotein receptor, LDLR) na powierzchni hepatocytów, zapobiegając rozkładowi LDLR przy udziale PCSK9. Zwiększenie gęstości występowania receptorów LDL wiąże się ze zmniejszeniem stężenia frakcji LDL cholesterolu w surowicy krwi.
1 wstrzykiwacz półautomatyczny napełniony zawiera 140 mg ewolokumabu w 1 ml roztworu. Preparat jest ludzkim przeciwciałem monoklonalnym klasy IgG2 wytwarzanym w komórkach jajnika chomika chińskiego (CHO) metodą rekombinacji DNA.